|
Reprinted with permission from Review of Ophthalmology. Kristine Morrell, Managing Editor, kmorrill@chilton.net
Robert W. Nickells, PhD
Department of Ophthalmology and Visual Sciences
University of Wisconsin
Madison, WI 53792
In North America, glaucoma in one form or another may affect more than 15 million persons (1). This disease is characterized by the progressive loss of retinal ganglion cells and axons that results in the excavation of the optic disc and the development of visual field defects beginning in the mid-peripheral retina. Of the known risk factors for glaucoma, an increased intraocular pressure (IOP) is most highly correlated with glaucomatous damage. Because of this correlation, the primary treatment of glaucoma is to lower the IOP. What physicians hope to accomplish by this is to reduce the rate of ganglion cell loss, and perhaps eliminate it altogether. Sometimes, this treatment appears to be effective, at least temporarily. Still, we are uncertain how effective pressure-management therapy is and it is hoped that on-going clinical trials like the Ocular Hypertension Study will help address this uncertainty.
With all this doubt about an effective glaucoma treatment, many of us have been asking if there are other treatments for this disease. Currently the answer to this question is no, but with recent scientific advances being made toward treating other neurodegenerative diseases, combined with an increased understanding of glaucoma, this answer could soon change to yes. These potential new treatments are directed at blocking neuronal death. There are a variety of different agents that seem to be able to do the job, many of them active on different facets of the various molecular pathways leading to cell death. Collectively, these agents are termed "neuroprotectants."
The Three Stages of Glaucoma
In order to appreciate how neuroprotectants may be used to treat glaucoma, I like to think of this disease as having three separate stages that are intimately linked together like a row of dominos (Figure 1). The first stage is the primary event, which for many people is probably a defect in the production/outflow of aqueous fluid leading to an increase in IOP. In susceptible individuals, this increased pressure leads to Stage 2, which I call the "damaging stimulus." Basically, an increased IOP leads to molecular changes in the retinal ganglion cells and surrounding tissues that activate Stage 3, which is the actual death of the ganglion cells. If we look at the progression of glaucoma as a row of falling dominos, then we can view pressure management therapy as being akin to removing some of the dominos early in the chain. At face value, this is a terrific strategy because if done early enough we can block the progression of this chain reaction. Unfortunately, we really have no good way of knowing if the chain reaction has already passed this critical point of treatment and this is probably why pressure management therapy is associated with variable success. The advent of new neuroprotective agents, however, means that it may soon be possible to start taking dominos out further down the chain, and at multiple sites, thus maximizing our chances of stopping the chain reaction before the irreversible death of the cells.
Stage 2 of Glaucoma: "The Damaging Stimulus"
In reality, it is probably more appropriate to refer to Stage 2 as having two "damaging stimuli" because basic research points to at least two reasonable mechanisms that can activate ganglion cell death in glaucoma. The first of these is neurotrophin deprivation. Neurotrophins are small peptides that are grouped in a class of molecules that also includes growth factors and neurotrophic factors. Basically, these small peptides interact with cell surface receptors that usually contain protein kinase domains. Activation of these receptors elicits a cascade of events that control cell growth and stasis. The survival of all neurons, including ganglion cells, is absolutely dependent on getting a fresh supply of neurotrophins on a regular basis. The neurotrophins themselves actually come from other cells that a neuron contacts suggesting these molecules act as a signaling mechanism constantly updating the status of neuronal connections. Retinal ganglion cells get their supply of neurotrophins from other neurons in the lateral geniculate nucleus or superior colliculus of the brain. Evidence has now shown that ganglion cells are dependent on a variety of neurotrophins, but primarily on one called brain-derived neurotrophic factor (BDNF). An adult ganglion cell takes up secreted BDNF from its respective target neuron and transports it along its axon back to the cell body in the retina. Glaucoma is now thought to block this retrograde flow of BDNF by blocking axoplasmic transport at the site of the lamina cribrosa (see reference (2) for review). It is not precisely known how long a ganglion cell can survive without its BDNF supply, but tests conducted in culture suggest that it is just a matter of days. One obvious neuroprotective strategy that is being considered for glaucoma treatment is to provide a different source of BDNF for the ganglion cells. This idea has already been tested successfully in rats that have had their optic nerves axotomized. Intravitreal injections of exogenous BDNF, and other growth factors, greatly reduces the rate of ganglion cell death after axotomy (3-5). The use of neurotrophins may have one disadvantage as a therapeutic agent, however. Early clinical trials showed that several molecules in this class produced unwanted side-effects when applied systemically (6).
The second damaging stimulus associated with glaucoma is the release of excitotoxins. These molecules are actually excitatory amino acids, such as glutamate, that are normally used by neurons as neurotransmitters. At high local concentrations, however, these normally benign molecules activate a highly toxic response in nearby cells (hence the derivation of the word "excitotoxin"). Like neurotrophins, excitotoxins interact with receptors on the cell surface. There are three sub-types of glutamate receptors found on neurons, but the one that appears to be the biggest player in the excitotoxic effect is the N-methyl-D-aspartate (NMDA) receptor (Figure 2). Glutamate binds to the receptor causing a channel to open and sodium and calcium to enter the cell. At very high concentrations of glutamate, this receptor is hyperstimulated and allows an excess of calcium to cross the cell membrane. This increase in intracellular calcium is thought to trigger a series of events including the activation of the enzyme Nitric Oxide Synthase (which creates the free radical form of nitric oxide) and the generation of reactive oxygen species (for review see reference (2)). Ultimately, these toxic substances do enough damage to the cell to bring about its demise.
Retinal ganglion cells are exquisitely sensitive to the excitotoxic effects of glutamate (Figure 3). Recently, elevated levels of glutamate were detected in the vitreous of both human glaucoma patients and monkeys with experimental glaucoma (7). Although the actual levels found in humans were small, similar concentrations were enough to cause the progressive death of ganglion cells in a rat model of chronic glutamate exposure (8). Currently, it is not known what stimulates the increase in glutamate in glaucoma, although neuronal ischemia is a possibility. It is also possible that any kind of damage, even neurotrophic deprivation, may elicit glutamate release. What is clear is that there are many ways to modulates the cellular response to glutamate, including drugs that block glutamate release and drugs that interact with the NMDA receptor to block either the interaction of the receptor and glutamate or affect the channel directly (a summary of some of these drugs is shown in Table 1).
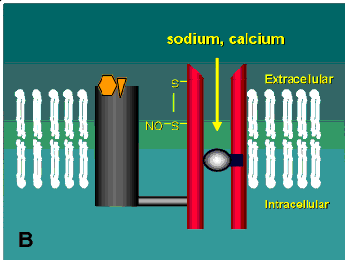
One of the most intriguing classes of drugs now being tested in neurodegenerative diseases is the open-channel blockers. These drugs actually enter and bind to the inside of the channel thus blocking the entry of calcium into the cell (Figure 2B). Some of these drugs are not suitable for therapeutic use because they tend to cause all sorts of neuropsychiatric side-effects. Angel dust (phencyclidine), for example, is an open-channel blocker. Others, such as dizocilpine, spend too long in the channel once it enters (dizocilpine has a half-time in the channel of more that an hour). These effects apparently stem from the fact that many neurons have NMDA receptors that are needed for normal function and systemic delivery of a drug affects all the channels not just the ones in cells undergoing an excitotoxic response. Alternatively, the short half-time channel blockers appear to be quite effective and some are currently being tested in Europe in clinical trials for other neurological disorders and AIDS-related dementia (see reference (9) for review). One of these compounds, memantine, has already been shown to be effective at blocking the excitotoxic response of retinal ganglion cells both in culture and in vivo (8, 10).
Stage 3 of Glaucoma: "Cell Death"
For a long time it seemed to glaucoma researchers that the end result of glaucoma was cell death and that was that. As a consequence, much of the research on glaucoma has been concentrated on understanding the "damaging stimuli" of this disease. Recently, however, we have realized that understanding how cells die is very important because, in some forms of cell death, there are lots of ways in which we can intervene in this process as well. Glaucoma, along with a variety of other important ocular diseases, all exhibit a form of cell death known as apoptosis (11). Apoptosis is actually a type of programmed cell death that is actively used by cells during development and in tissue homeostasis. As this name implies, it is a cell-autonomous phenomenon in that the death of the cell is already pre-programmed in its genes. When it receives the correct signal, the cell executes the program which then causes it to commit suicide. In the normal remodeling of the ganglion cell layer that occurs in development, this signal is neurotrophin deprivation (see reference (2) for review). Glaucoma, it appears, may be a disease in which ganglion cells accidentally receive a normal developmental signal to begin apoptosis. This isn't the only thing that can stimulate apoptosis of ganglion cells, however, because recent studies indicate that chronic exposure to glutamate can do the same thing.
The molecular pathways associated with apoptosis are very complex, and not all cells seem to utilize the same pathways although some are highly conserved across very diverse phyla. The basic molecular events of apoptosis start with a signal that probably leads to minimal damage to the cell, such as in the case of excitotoxin exposure. This signal is not well understood, but it may be an increase in oxygen free radicals which go around chemically reacting with almost everything in the cell. This elicits a chain of events that appear to be regulated by the tumor suppressor gene, p53. This protein can alter the expression of several other genes that eventually leads to the activation of catabolic enzymes, particularly those that degrade the cellular DNA (nucleases) and protein components (proteases). There are several drugs that are proving extremely useful at blocking apoptosis. Many of these are grouped as anti-oxidants, which may function to block the initial stages of the apoptotic program. One class of compounds that block lipid peroxidation (collectively known as "Lazaroids") has been shown to delay retinal ganglion cell death in culture (12). Other drugs exist that block the actions of the proteases (protease inhibitors). Currently, these drugs are still being characterized and tested in the laboratory (13). It is now clear that different members of a family of apoptosis-related proteases are active in different cells and future treatments may first require identifying which are active in target cells. Given the recent success of protease inhibitors in treating AIDS (14), however, this class of molecules may prove to be very powerful neuroprotectants.
In summary, we appear to be at the beginning of an exciting time for the treatment of glaucoma. Progress in research has shown us that both the activation and the process of cell death in this disease are areas that can be treated. We should also benefit from the several clinical trials that are ongoing to test multiple neuroprotectants in the treatment of other neurodegenerative disorders like stroke, Alzheimer's disease and amyotrophic lateral sclerosis. Not only do these diseases appear to be somewhat similar to glaucoma, but effective , FDA-approved drugs, may be available in the near future. Ultimately, the treatment of glaucoma may be a multi-step process, starting with pressure-management therapy, followed by neuroprotectants aimed at preventing the activation of ganglion cell apoptosis, and ending with neuroprotectants that block some aspect of the apoptotic program. The more dominos that we can get our hands on, the more effective our treatment of this disease will become.
References
- Quigley HA. Open-angle glaucoma. New Engl. J. Med. 1993;328:1097-1106.
- Nickells RW. Retinal ganglion cell death in glaucoma: the how, the why, and the maybe. J. Glaucoma 1996;5:345-356.
- Sievers J, Hausmann B, Unsicker K, Berry M. Fibroblast growth factors promote the survival of adult rat retinal ganglion cells after transection of the optic nerve. Neurosci. Lett. 1987;76:157-162.
- Mey J, Thanos S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993;602:304-317.
- Mansour-Robaey S, Clarke DB, Wang Y-C, Bray GM, Aguayo AJ. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1994;91:1632-1636.
- Barinaga M. Neurotrophic factors enter the clinic. Science 1994;264:772-774.
- Dreyer EB, Zurakowski D, Schumer RA, Podos SM, Lipton SA. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch. Ophthalmol. 1996;114:299-305.
- Vorwerk CK, Lipton SA, Zurakowski D, Hyman BT, Sabel BA, Dreyer EB. Chronic low-dose glutamate is toxic to retinal ganglion cells. Toxicity blocked by memantine. Invest. Ophthalmol. Vis. Sci. 1996;37:1618-1624.
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. New Engl. J. Med. 1994;330:613-622.
- Chen H-S, Pellegrini JW, Aggarwal SK, et al. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J. Neurosci. 1992;12:4427-4436.
- Nickells RW, Zack DJ. Apoptosis in ocular disease: A molecular overview. Ophthalmic Genetics 1996;(in press).
- Levin LA, Clark JA, Johns LK. Effect of lipid peroxidation inhibition on retinal ganglion cell death. Invest. Ophthalmol. Vis. Sci. 1996;37:2744-2749.
- Lotem J, Sachs L. Differential suppression by protease inhibitors and cytokines of apoptosis induced by wild-type p53 and cytotoxic agents. Proc. Natl. Acad. Sci. USA 1996;93:12507-12512.
- Cohen J. Protease inhibitors: A tale of two companies. Science 1996;272:1882-1883.
Figure Legends
Figure 1.
In susceptible people, the three stages of glaucoma are linked like a row of dominos. Initiation of the first stage results in each stage progressing to the next like a chain reaction. Treatments for glaucoma are like taking dominos out of the chain, thus blocking this progression.
Back |
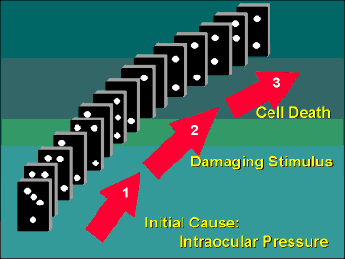 |
Figure 2.
A schematic of the NMDA receptor. (A) Glutamate (glycine acts as a co-agonist) binds to one region of the receptor and causes the channel to open allowing sodium and calcium to enter into the cell. (B) Open-channel blockers act by entering the channel and blocking the flow of these ions. This receptor has several other potential modulatory sites including a redox site that may react with oxidized nitric oxide leading to the formation of a disulphide bridge on the channel (see Table 1). Diagram modified from reference (9).
Back |
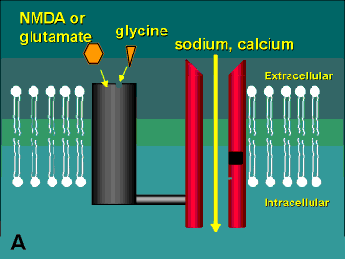 |
Figure 3.
An example of the effects of excitotoxins on retinal ganglion cells. In this experimental situation, normal saline was injected into the vitreous of the left eye of a mouse (A) and saline containing excitotoxins was injected into the right eye (B). After 2 days, the retinas were processed for histology and stained with a fluorescent nuclear dye. Only ganglion cells are affected by exposure to excitotoxins. (GCL=ganglion cell layer). Magnification X420.
Back |
 |
| Site of Action |
Compound |
Mode of Action |
Notes |
| Pre-NMDA Receptor |
| |
Riluzole |
Attenuates glutamate release |
FDA-approved for treating amyotrophic lateral sclerosis (ALS) |
| |
Lifarazine |
Attenuates glutamate release |
Reduces neuronal damage in animal models of stroke |
| NMDA Receptor |
| |
CGS19755 |
glutamate binding-site antagonist |
|
| |
Felbamate |
glycine binding-site antagonist |
FDA-approved as an anticonvulsant; currently being
tested on some forms of epilepsy |
| |
Magnesium |
Open-channel blocker |
Very short half-time in channel (may be too short to
be effective?) |
| |
Memantine |
Open-channel blocker |
Effective neuroprotectant of retinal ganglion cells in
animal models |
| |
Nitroglycerin |
Redox site |
Provides a source of oxidized nitric oxide; reduces
neuronal damage in animal models of stroke |
|